18017847121

《Advanced science》是Wiley出版集團旗下的一本開放獲取(Open Access)綜合性學術期刊,于2014年創刊。在中科院最新升級版分區表中,大類學科為材料科學1區,小類學科中化學綜合為1區、納米科技為2區、材料科學綜合為1區,屬于TOP期刊。《Advanced science》是一本跨學科開放獲取期刊,涵蓋材料科學、物理和化學、醫學和生命科學以及工程學等領域,主要發表這些領域的yi流基礎和應用研究成果。
出版周期:12 issues/year;
影響因子:14.1;
ISSN:2198-3844;
發文量:3290篇/年;
審稿速度:平均12周;
版面費:USD 5270.00;

一、研究背景與目標
HCC 是全球致死率最高的癌癥之一,傳統肝切除術后復發率高,而PD-1/PD-L1免疫治療響應率僅 15-30%。其核心瓶頸在于腫瘤微環境(TME)的免疫抑制,尤其是TAMs的異常極化(向M2型轉化)會抑制T細胞功能。本研究旨在探索TAMs中NUPR1的作用及機制。
二、關鍵技術總結
研究通過多種實驗技術解析NUPR1在肝癌腫瘤相關巨噬細胞(TAMs)中的作用及機制,具體如下:
1、組學分析
單細胞RNA測序(scRNA-seq):對人及小鼠肝癌組織的單細胞轉錄組數據(GSE149614、GSE232182等數據集)進行整合分析,鑒定NUPR1在TAMs中的特異性高表達,區分NUPR1高/低表達巨噬細胞亞群,并通過基因集富集分析(GSEA)揭示其功能差異(如免疫抑制通路富集)。利用UMAP可視化細胞聚類,結合Harmony算法校正批次效應,確保跨數據集分析的一致。
空間轉錄組(ST)分析:對肝癌組織空間轉錄組數據(GSE238264)進行分析,驗證NUPR1與巨噬細胞標志物CD68 在腫瘤區域的共定位,明確其在腫瘤微環境中的空間分布特征。
bulk RNA-seq與多數據庫整合:整合 TCGA、GEO、ICGC 等數據庫的bulk RNA-seq數據,構建NUPR1?巨噬細胞特征基因集,關聯其表達與患者預后及免疫治療響應
2、巨噬細胞極化和共培養模型
通過腫瘤條件培養基(TCM)誘導THP-1細胞或骨髓來源巨噬細胞(BMDMs)向TAMs極化,結合NUPR1抑制劑(TFP-2HCL、ZZW-115-2HCL)或shRNA敲低技術,研究NUPR1 對巨噬細胞表型的調控
建立巨噬細胞與CD8?T細胞共培養體系,通過流式細胞術檢測T細胞功能標志物(IFN-γ、Gzmb、PD-1),評估NUPR1對T細胞耗竭的影響
3、基因與蛋白水平檢測
定量實時PCR(qRT-PCR):檢測巨噬細胞中M1/M2 標志物(如iNOS、CD86、CD206、ARG1)及NUPR1、PD-L1等基因的表達水平,驗證NUPR1對極化表型的調控。
Western blotting:分析 ERK、JNK等MAPK通路蛋白的磷酸化水平,以及組蛋白乳酸化(H3K18la)、NUPR1等蛋白表達,揭示分子調控機制。
染色質免疫沉淀(ChIP):通過ChIP-qPCR驗證乳酸誘導的H3K18la在NUPR1啟動子區域的富集,證實組蛋白乳酸化對NUPR1轉錄的激活作用
4、動物模型與體內實驗技術
皮下移植瘤模型:將Hepa1-6細胞接種于C57BL/6小鼠腋窩,評估NUPR1抑制劑對腫瘤生長、巨噬細胞極化及T細胞浸潤的影響。
原位肝癌模型:通過hydrodynamic尾靜脈注射攜帶c-Myc和Ctnnb-N90質粒的溶液誘導小鼠肝臟自發性腫瘤,模擬更接近臨床的腫瘤微環境。
巨噬細胞轉移實驗:將經TFP-2HCL預處理的BMDMs瘤內注射,驗證靶向巨噬細胞NUPR1對腫瘤生長的抑制作用。
免疫治療聯合實驗:在小鼠模型中聯合使用NUPR1抑制劑(TFP-2HCL或ZZW-115-2HCL)與PD-1單克隆抗體,通過腫瘤體積監測、流式細胞術及IHC染色,評估聯合治療對免疫微環境的改善效果。
5、病理與影像技術
多重免疫熒光(mIF):檢測腫瘤組織中NUPR1?CD68?巨噬細胞與CD8?T細胞的共定位及數量變化,關聯免疫治療響應。
免疫組化(IHC):分析腫瘤組織中CD206、iNOS、CD8、PD-1等標志物的表達,評估巨噬細胞極化和T細胞浸潤狀態。
影像評估:通過磁共振成像(MRI)監測肝癌患者免疫治療前后的腫瘤變化,關聯NUPR1?巨噬細胞水平與治療響應。
三、主要研究結果
1、NUPR1在HCC腫瘤相關巨噬細胞(TAMs)中高表達且參與HCC進展
通過多維度分析揭示NUPR1在肝癌(HCC)腫瘤相關巨噬細胞(TAMs)中的表達特征及臨床意義。scRNA-seq數據顯示,與正常肝組織相比,HCC腫瘤組織中巨噬細胞比例顯著升高,T/NK細胞比例減少。差異基因分析發現NUPR1是腫瘤與正常組織巨噬細胞的核心差異基因,且在腫瘤巨噬細胞中高表達。跨物種驗證顯示,小鼠HCC模型中NUPR1同樣主要在巨噬細胞中表達。空間轉錄組證實NUPR1與CD68在腫瘤區域共定位,且隨巨噬細胞浸潤腫瘤時間動態上調。臨床數據表明,NUPR1?巨噬細胞特征基因在腫瘤組織中富集,其高表達與HCC患者更低的總生存率、更高的復發率顯著相關,是術后預后的獨立預測因子。且僅巨噬細胞中NUPR1表達與預后相關,整體NUPR1表達分層無顯著差異,強調其細胞特異性作用。結果明確了NUPR1在HCC TAMs中特異性高表達的特征,并證實其作為不良預后標志物的臨床價值。
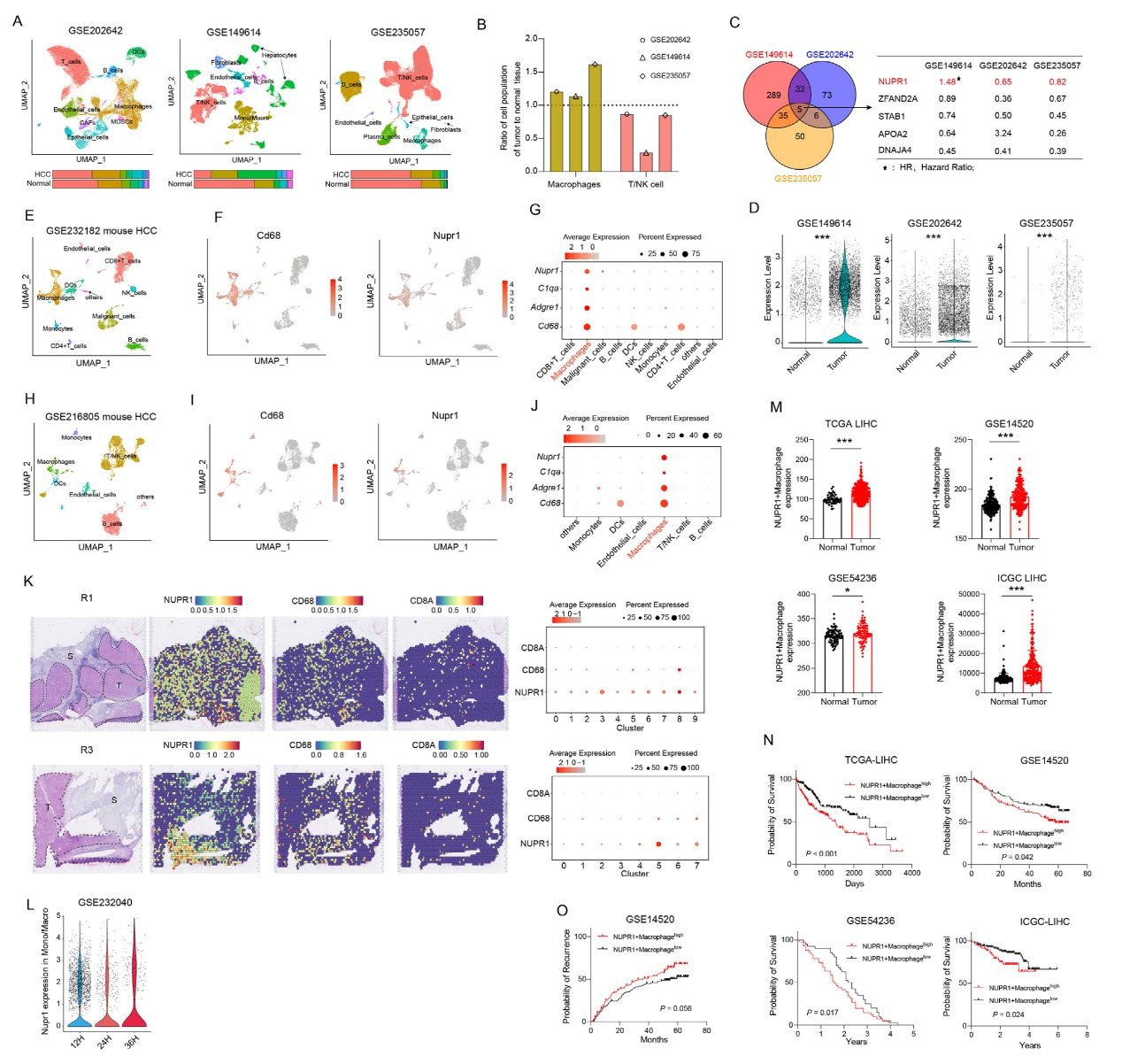
圖1、NUPR1在HCC TAMs中高表達
2、NUPR1對巨噬細胞表型及功能的調控作用
通過對NUPR1高/低表達巨噬細胞的分析,發現NUPR1高表達巨噬細胞富集免疫抑制通路,而低表達組富集免疫激活通路。表型上,NUPR1高表達巨噬細胞呈M2型特征(高表達ARG1、TREM2等),并高表達PD-L1、SIRPA等免疫檢查點;低表達組則呈M1型特征(高表達 CD80、IL1B等)。NUPR1抑制劑TFP-2HCL處理后,THP1細胞和BMDMs的M1標志物(iNOS、CD86)表達升高,M2 標志物(CD206、ARG1)表達降低,巨噬細胞形態從M2 樣紡錘形轉向M1樣圓形。臨床樣本mIF染色顯示腫瘤組織中NUPR1?CD68?細胞更多,且其低表達患者預后更好,證實NUPR1在巨噬細胞極化及預后中的關鍵作用。
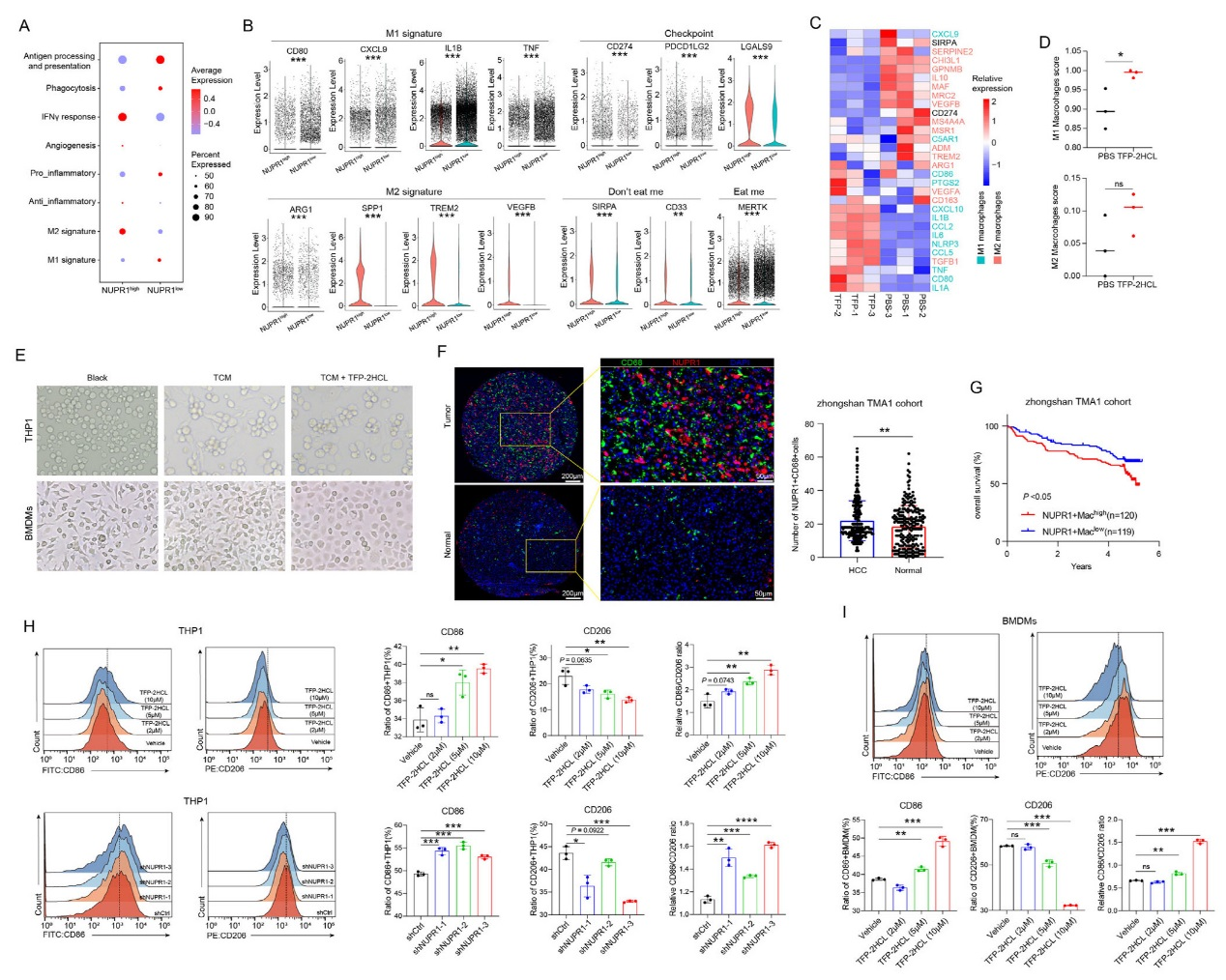
圖2、NUPR1在巨噬細胞中維持免疫抑制表型
3、NUPR1通過抑制MAPK通路調控巨噬細胞表型轉換的機制
基因集富集分析(GSEA)顯示,NUPR1低表達巨噬細胞中MAPK信號通路顯著富集,其通路評分顯著高于NUPR1高表達組。UMAP可視化進一步證實,NUPR1高表達區域對應更低的MAPK通路活性,且NUPR1表達與MAPK通路評分呈顯著負相關。KEGG分析顯示,NUPR1抑制劑TFP-2HCL處理的THP1細胞中MAPK通路顯著激活,ssGSEA驗證其通路評分升高。Western blot結果表明,TFP-2HCL處理或NUPR1敲低后,THP1細胞和骨髓來源巨噬細胞(BMDMs)中ERK和JNK磷酸化水平顯著增加,但p38磷酸化無明顯變化。功能實驗顯示,TFP-2HCL可誘導巨噬細胞從M2型向M1型極化,而ERK抑制劑(FR180204)或JNK抑制劑(JNK-IN-8)可逆轉這一效應。研究結果證實NUPR1通過抑制ERK和JNK磷酸化抑制MAPK通路,進而維持巨噬細胞的免疫抑制表型,而靶向NUPR1可激活ERK/JNK通路,促進M1型極化。
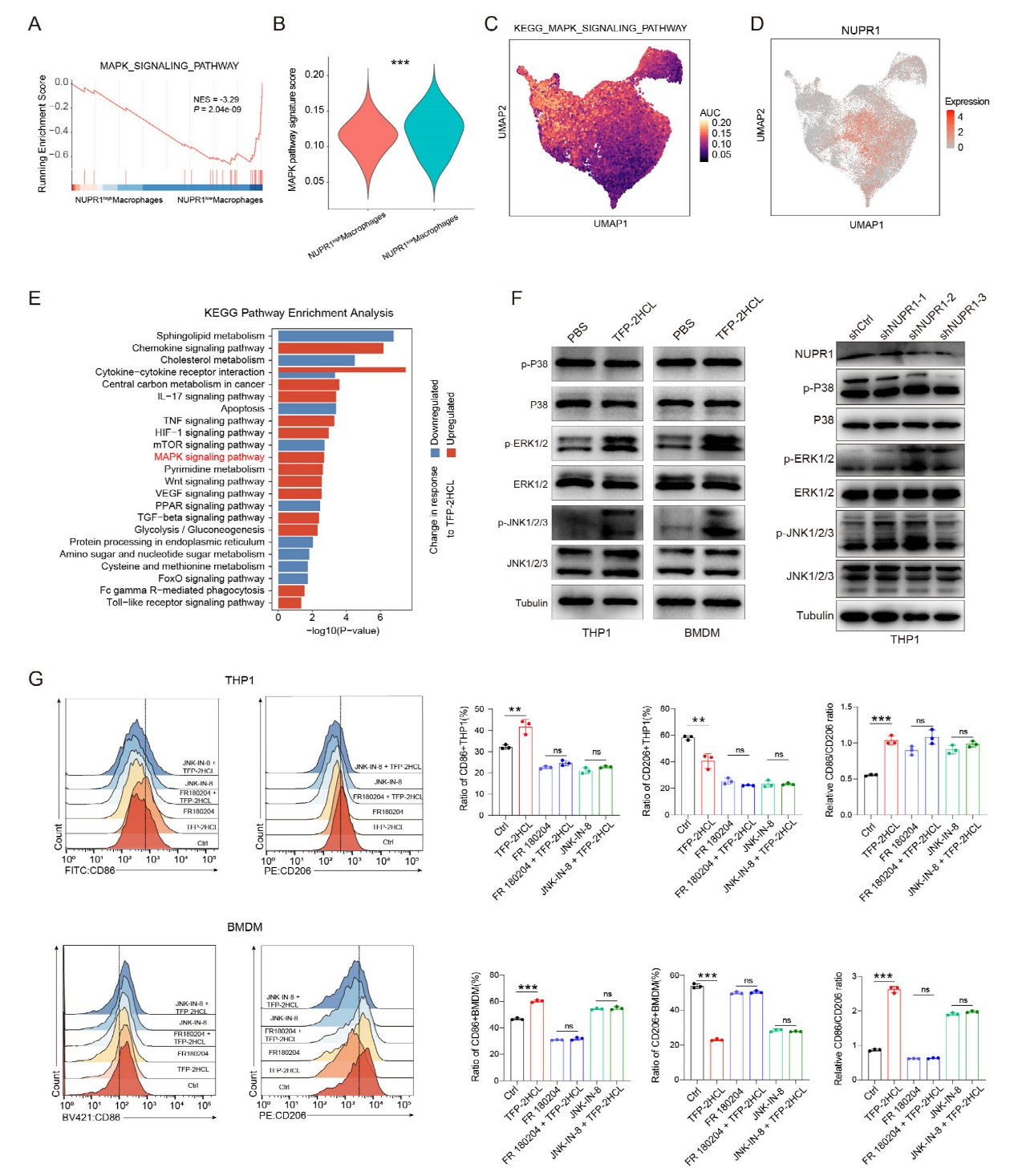
圖3、NUPR1通過ERK和JNK通路促進巨噬細胞表型轉換
4、NUPR1?巨噬細胞對 CD8?T 細胞功能的影響及機制
scRNA-seq分析顯示,NUPR1高/低表達組的T/NK細胞比例無顯著差異,但基因表達特征不同:NUPR1高表達組中CD8?T細胞的細胞毒性基因低表達,耗竭基因高表達;低表達組則progenitor耗竭基因富集。signature評分顯示,NUPR1高表達組CD8?T細胞耗竭評分升高,細胞毒性和progenitor耗竭評分降低。TCGA數據證實,NUPR1?巨噬細胞與耗竭CD8?T細胞呈強正相關。共培養實驗中,TFP-2HCL處理的巨噬細胞可上調CD8?T細胞的IFN-γ、Gzmb表達,減少PD-1表達,逆轉T細胞耗竭。而加入ERK抑制劑(FR180204)或JNK 抑制劑(JNK-IN-8)后,TFP-2HCL對CD8?T細胞的調控作用被消除。NUPR1?巨噬細胞通過抑制ERK/JNK通路促進CD8?T細胞耗竭,靶向NUPR1可通過激活該通路改善T細胞功能。
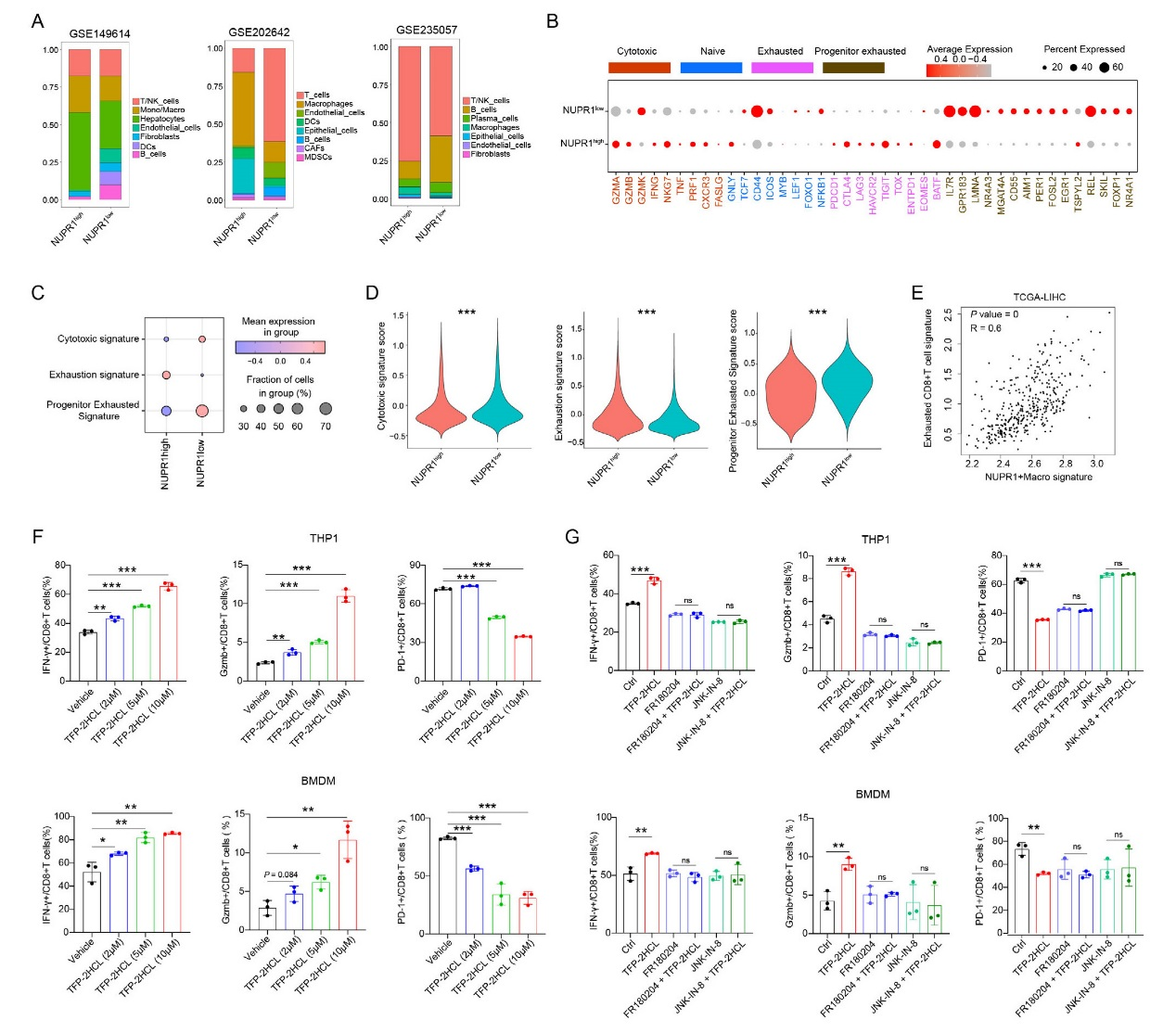
圖4、巨噬細胞中的NUPR1通過ERK和JNK通路促進耗竭性CD8+ T細胞的耗竭
5、靶向NUPR1對肝癌(HCC)的治療效果及與PD-1抑制劑的協同作用
在Hepa1-6皮下移植瘤模型中,NUPR1抑制劑TFP-2HCL處理顯著縮小腫瘤體積、降低重量,腫瘤組織中M2標志物CD206減少,M1標志物iNOS增加,CD8?T細胞浸潤增多且 PD-1表達降低。hydrodynamic尾靜脈注射誘導的自發性原位肝癌模型中,TFP-2HCL同樣顯著減輕肝臟腫瘤負荷,IHC染色顯示類似的免疫微環境改善。聯合治療實驗中,TFP-2HCL 與抗PD-1抗體聯用較單藥更有效抑制腫瘤生長,促進巨噬細胞M1極化,增加IFN-γ?、Gzmb?CD8?T細胞,減少PD-1?CD8?T細胞。
低神經毒性的NUPR1抑制劑ZZW-115-2HCL單藥或與PD-1抑制劑聯用,在自發性模型中也顯著增強抗腫瘤效率。巨噬細胞轉移實驗證實,TFP-2HCL預處理的巨噬細胞可抑制腫瘤生長。結果表明,靶向 NUPR1能重塑免疫微環境,與PD-1抑制劑協同增強HCC治療效果。
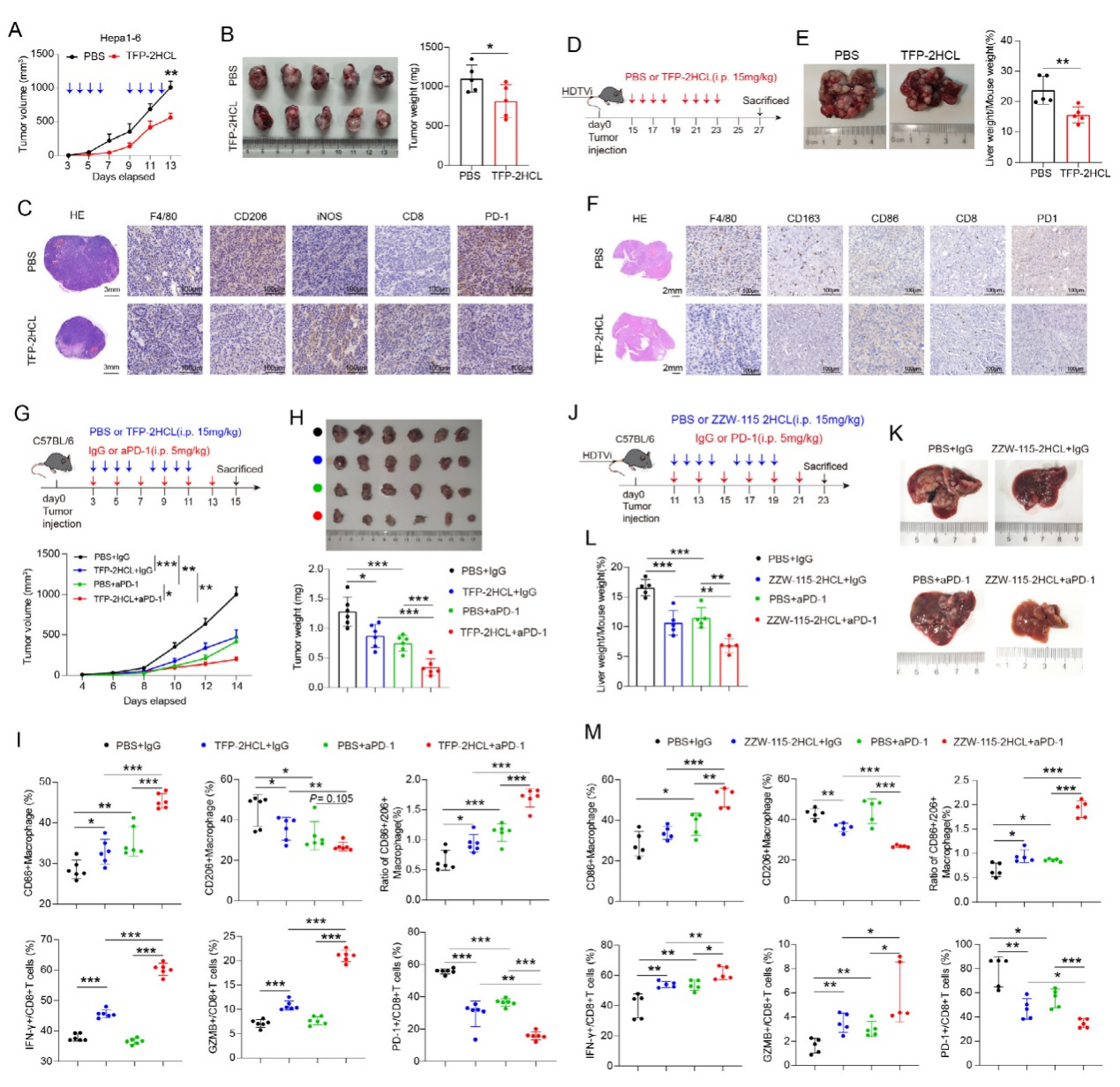
圖5、抑制NUPR1可增強抗PD-1治療在HCC中的體內療效
6、腫瘤來源乳酸通過組蛋白乳酸化上調巨噬細胞NUPR1的機制。
Western blot 顯示,與腫瘤條件培養基(TCM)共培養后,THP1細胞和骨髓來源巨噬細胞(BMDMs)中 NUPR1 表達顯著升高,且乳酸處理可濃度依賴性增強NUPR1表達。GSE149614 數據集分析顯示,NUPR1高表達巨噬細胞對應的腫瘤細胞 glycolysis評分更高,糖酵解相關基因表達上調。TCGA數據證實,NUPR1?巨噬細胞與乳酸轉運體SLC16A3呈強正相關。機制上,TCM或乳酸處理可增加巨噬細胞中組蛋白H3K18乳酸化水平,同時抑制 ERK和JNK磷酸化。代謝干預實驗顯示,腫瘤細胞經糖酵解抑制劑(Oxamate、2-DG)處理后,其條件培養基誘導 NUPR1表達的能力減弱;而糖酵解促進劑Rotenone則增強該效應。ChIP-qPCR證實,TCM或乳酸可通過H3K18乳酸化富集NUPR1啟動子區域,激活其轉錄。綜上,腫瘤細胞分泌的乳酸通過組蛋白H3K18乳酸化上調巨噬細胞NUPR1表達,抑制ERK/JNK通路。
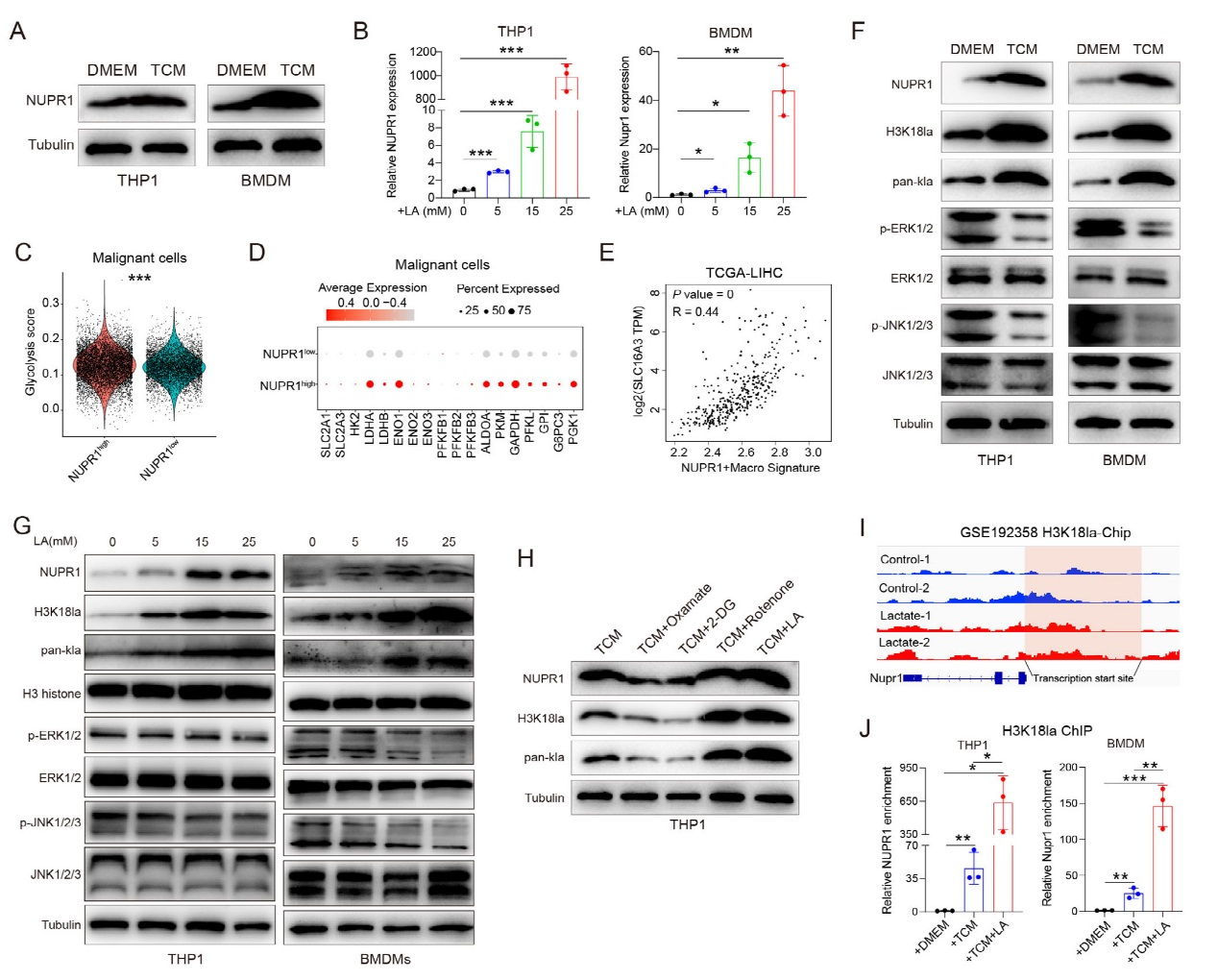
圖6、腫瘤細胞通過H3K18賴氨酸促進巨噬細胞中NUPR1的表達。
7、NUPR1?巨噬細胞與免疫治療耐藥的關聯
MRI影像顯示HCC患者PD-1單抗治療的響應差異。mIF染色及量化分析表明,非響應者(42 例)腫瘤中NUPR1?CD68?細胞數量顯著高于響應者(33 例),且非響應者中CD68?CD206?NUPR1?(M2型)細胞比例更高。Kaplan-Meier曲線顯示,高NUPR1?CD68?水平的HCC患者總生存期更短。跨癌種分析發現,NSCLC和黑色素瘤中NUPR1主要表達于巨噬細胞,非響應者中其表達顯著升高。多數據集驗證顯示NUPR1?巨噬細胞高表達與耐藥及不良預后相關,結果表明,乳酸通過H3K18乳酸化上調NUPR1并導致免疫抑制。
圖7、NUPR1+巨噬細胞介導對免疫療法的抗性
四、全文結論
本研究揭示,核蛋白 1(NUPR1)在肝細胞癌(HCC)腫瘤相關巨噬細胞(TAMs)中扮演關鍵角色。NUPR1在TAMs中高表達,與患者不良預后緊密相關,其可促進M2巨噬細胞極化,上調免疫檢查點分子如PD-L1和SIRPA的表達,進而抑制CD8?T細胞功能,營造免疫抑制性腫瘤微環境,降低PD-1阻斷治療的響應率。機制上,腫瘤來源的乳酸誘導組蛋白 H3K18乳酸化,上調NUPR1轉錄,NUPR1則通過抑制 ERK 和JNK信號通路發揮免疫抑制作用。通過藥理學手段抑制NUPR1,可有效逆轉M2極化,增強CD8?T細胞功能。在臨床前模型中,抑制NUPR1與PD-1單抗聯合使用,顯著抑制腫瘤生長。此外,NUPR1?TAMs可作為預測免疫治療響應的生物標志物,為HCC免疫治療耐藥問題提供了新的解決思路,NUPR1有望成為提升HCC免疫治療療效的潛在靶點。
參考文獻:
Cai J, Zhang P, Cai Y, Zhu G, Chen S, Song L, Du J, Wang B, Dai W, Zhou J, Fan J, Yu Y, Dai Z. Lactylation-Driven NUPR1 Promotes Immunosuppression of Tumor-Infiltrating Macrophages in Hepatocellular Carcinoma. Adv Sci (Weinh). 2025 May;12(20): e2413095. doi: 10.1002/advs.202413095. Epub 2025 Apr 30. PMID: 40305758; PMCID: PMC12120759.
 歡迎來到
歡迎來到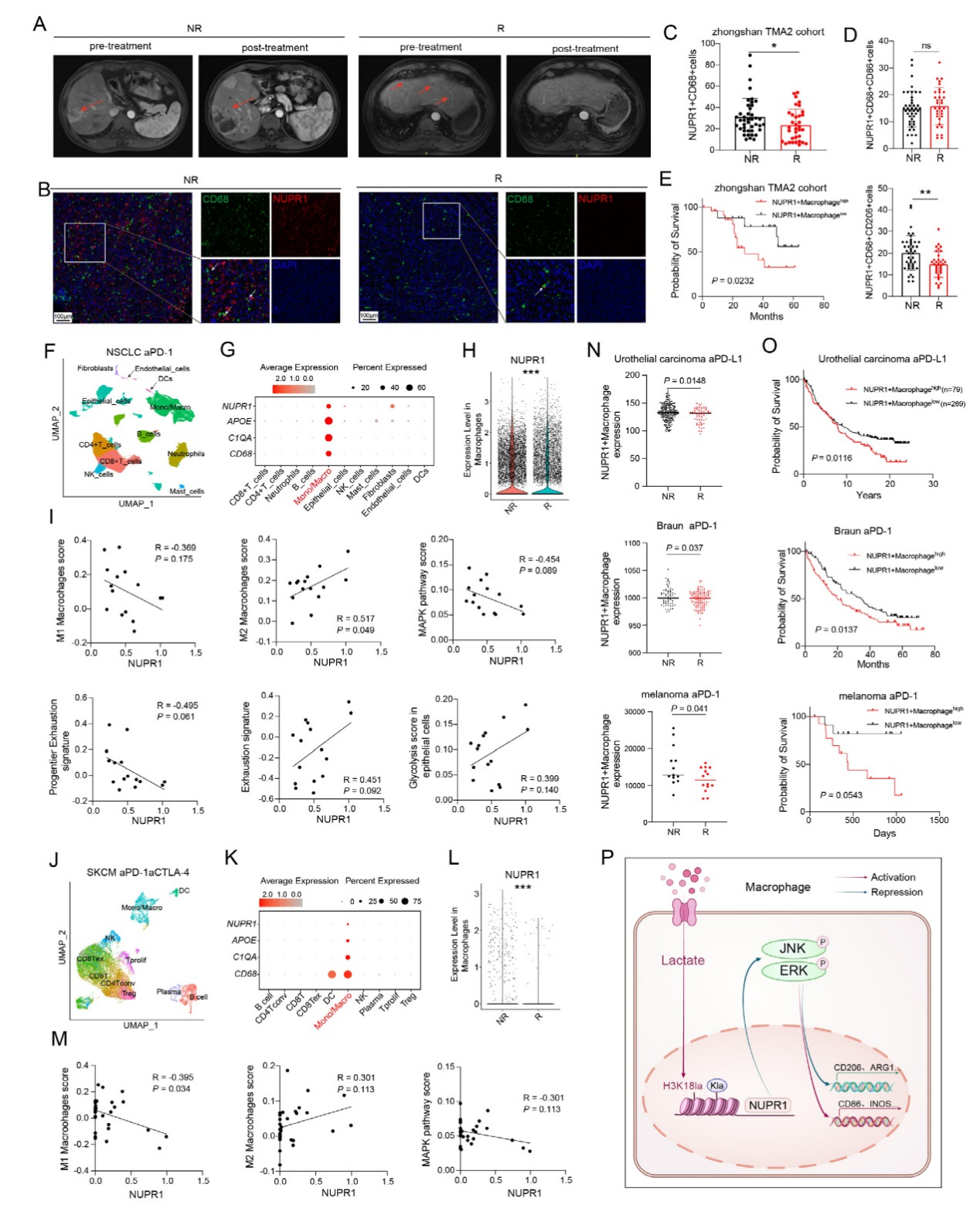
 聯系人:張
聯系人:張 地址:上海市長江南路180號長江軟件園B區B637室
地址:上海市長江南路180號長江軟件園B區B637室 郵箱:2844970554@qq.com
郵箱:2844970554@qq.com 傳真:
傳真: