18017847121

朋友們,今天要帶大家探索一篇雜志《Nature communications》
影響因子:15.7
期刊ISSN:2041-1723
分區:中科院1 區,且為 TOP 期刊。
發文量:10926/年(2024年)
自引率:3.40%
審稿速度:平均審稿周期約1個月
下面這篇文章就發表在《Nature communications》上:
文章標題:Macrophage-fibroblast JAK/STAT dependent crosstalk promotesliver metastatic outgrowth in pancreatic cancer
發表時間:2024 年
核心內容:研究發現胰腺癌肝轉移中,巨噬細胞通過分泌顆粒蛋白前體,與癌細胞分泌的白血病抑制因子協同激活成纖維細胞的 JAK/STAT3 信號,誘導肌成纖維細胞(myMAF)生成,其分泌的骨橋蛋白進一步塑造免疫抑制微環境,促進轉移,并且阻斷該通路可抑制轉移。
一、 研究背景
胰腺導管腺癌(PDAC)是一種高度致命的惡性腫瘤,其侵襲性轉移特性是導致患者預后極差的主要原因。大多數患者在確診時已出現不可切除的轉移性病灶,且即使手術切除后仍有超過60%的患者在兩年內發生肝轉移復發。轉移是一個多階段過程,涉及癌細胞從原發灶脫落、循環擴散和遠端器官定植,而這一過程的成功高度依賴于腫瘤微環境中基質細胞的支持,特別是巨噬細胞的作用。然而,目前對PDAC肝轉移中巨噬細胞的具體功能機制仍知之甚少。
該研究聚焦于PDAC最常見的轉移靶器官——肝臟,發現轉移灶中富含巨噬細胞和活化的肌成纖維細胞。研究揭示了巨噬細胞通過分泌顆粒蛋白前體激活肝星狀細胞(hStCs),進而誘導纖維化微環境形成,促進轉移性腫瘤生長的關鍵機制。這一發現不僅填bu了PDAC轉移機制的重要空白,還為開發靶向顆粒蛋白前體或巨噬細胞招募的新型治療策略提供了理論依據,有望改善PDAC患者的臨床預后。
二、 研究思路
首先研究人員基于PDAC易發生肝轉移但機制不明的臨床問題,通過小鼠模型和臨床樣本分析發現轉移灶中巨噬細胞(MAMs)顯著聚集并高表達顆粒蛋白前體;隨后通過實驗證實這些巨噬細胞通過分泌顆粒蛋白前體激活肝星狀細胞(hStCs),促使后者分泌骨膜蛋白等基質蛋白形成促轉移的纖維化微環境;進一步機制研究發現巨噬細胞招募依賴PI3Kγ信號通路,而阻斷該通路或敲除顆粒蛋白前體均可顯著抑制轉移;最終提出靶向巨噬細胞-顆粒蛋白前體-hStCs軸可能是治療PDAC肝轉移的新策略,為臨床轉化提供理論依據。
三、 研究結果
1. 轉移相關巨噬細胞調節轉移性PDAC中的成纖維細胞異質性
研究人員通過免疫熒光染色和Pdgfrb-GFP小鼠模型,發現PDAC肝轉移中PDGFRβ+間充質細胞與CD68+巨噬細胞增多;耗竭巨噬細胞后,轉移相關巨噬細胞減少65%,轉移負荷和肝纖維化降低;單細胞測序鑒定出了MAFs的四個亞群:vMAFs、myMAFs、iMAFs和cycMAFs,其中myMAFs占比43%,耗竭巨噬細胞后豐度下降至19%,且myMAFs、vMAFs和cycMAFs起源于肝星狀細胞,iMAFs起源于成纖維細胞。
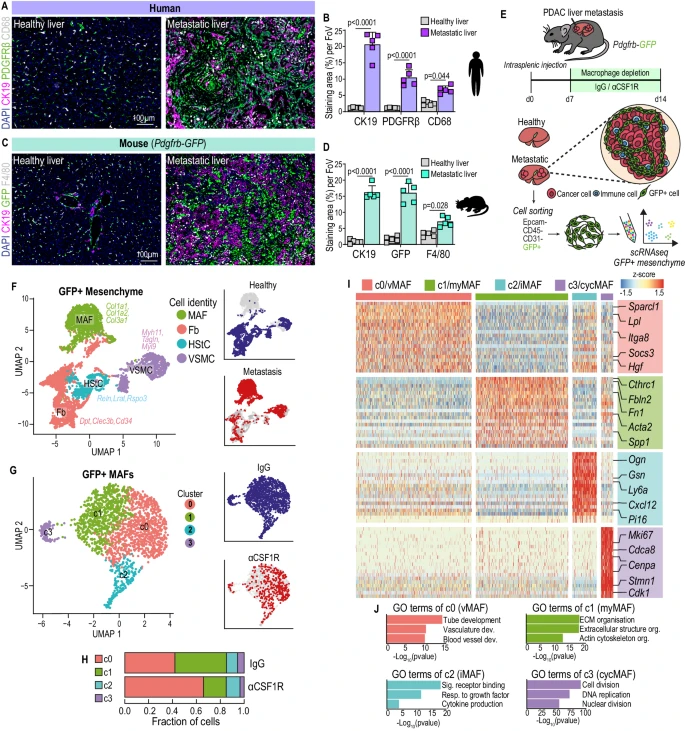
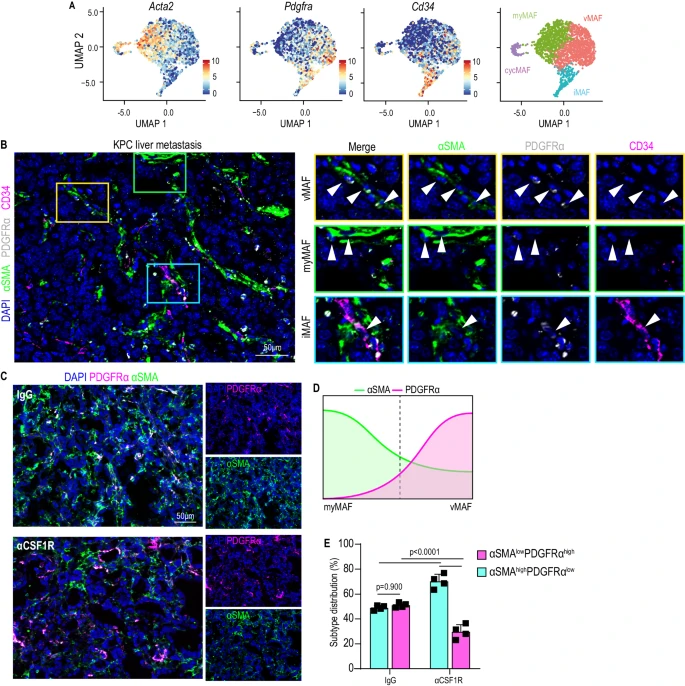
2. 轉錄和空間上多樣化的MAF群體在PDAC肝轉移中共同存在
研究人員通過免疫熒光染色驗證了vMAF、myMAF和iMAF三個MAF亞群在小鼠和人類PDAC肝轉移中的保守存在,利用α-SMA、PDGFRα和CD34的分級表達進行原位檢測,發現vMAFs位于腫瘤血管附近,myMAFs存在于血管化較少區域,且巨噬細胞耗竭會使vMAFs增多、myMAFs減少,將纖維化微環境從典型的富含膠原的促纖維增生狀態重塑為血管化逐漸增強的腫瘤微環境。
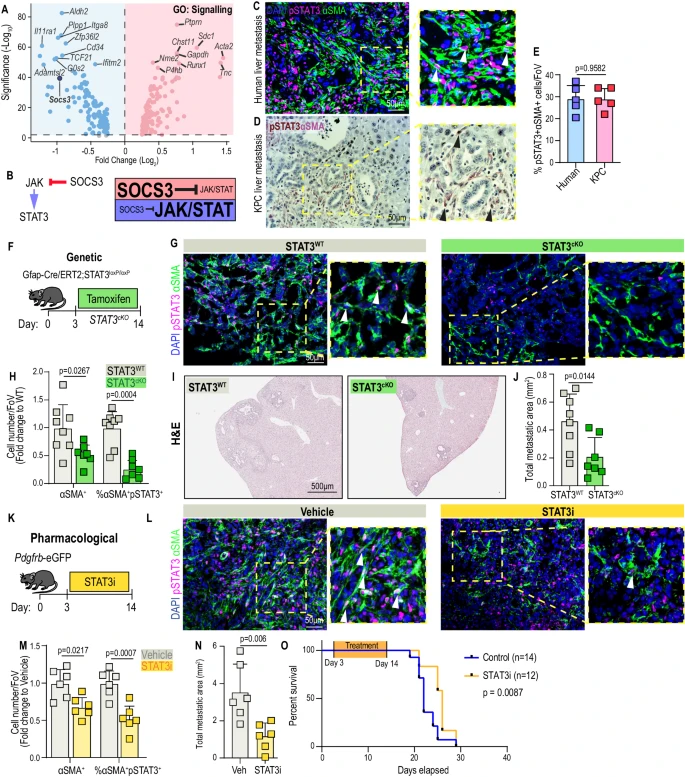
3. myMAFs 促進肝轉移,其促轉移功能依賴于 JAK/STAT 信號傳導通路的激活
研究人員前期研究發現富含膠原的ECM可促進癌癥進展,而myMAFs是其主要來源,且巨噬細胞耗竭能顯著減少myMAFs。研究人員通過進一步研究發現,JAK/STAT通路抑制因子Socs3在myMAFs中顯著下調,該通路在30%的αSMA+細胞中激活并主要富集于myMAFs。通過GFAP-STAT3敲除小鼠模型和水飛薊賓藥理抑制實驗證實,STAT3缺失或抑制可減少myMAF活化、膠原沉積及肝轉移負荷,延長荷瘤小鼠生存期,表明巨噬細胞通過JAK/STAT通路參與了促腫瘤性myMAFs積累,靶向該通路可阻斷其促轉移功能。
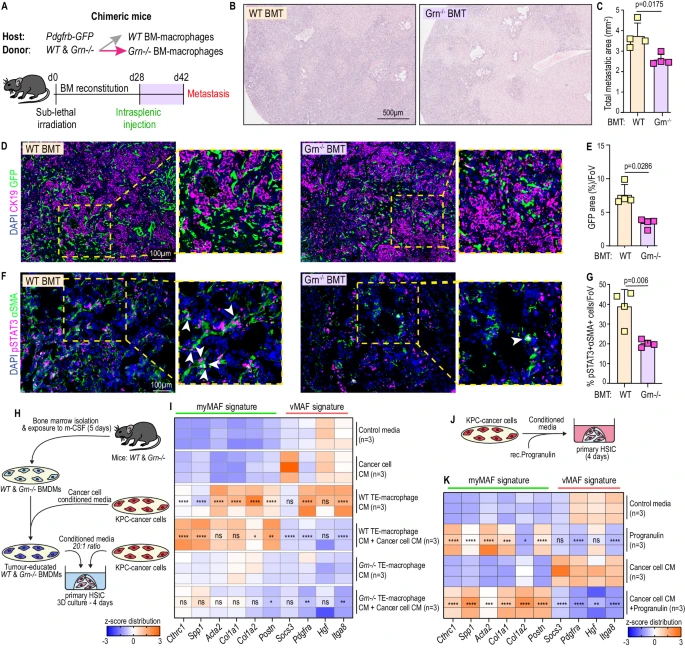
4. 顆粒蛋白前體與癌細胞衍生因子的共刺激可促進myMAF的活化
研究發現JAK/STAT是myMAF活化的關鍵通路,為探究巨噬細胞如何在肝星狀細胞中激活該通路,研究人員構建了攜帶野生型或缺乏顆粒蛋白前體(Grn?/?)骨髓的嵌合小鼠,發現Grn?/?骨髓小鼠轉移灶生長、纖維化程度及αSMA+細胞中pSTAT3+細胞比例均顯著降低;通過三維培養系統進一步證實,巨噬細胞分泌的顆粒蛋白前體與癌細胞來源因子協同作用,可誘導肝星狀細胞向myMAF表型轉化,結果表明,巨噬細胞來源的顆粒蛋白前體在myMAF活化中起到關鍵作用。
5. 中和癌細胞衍生的白血病抑制因子(LIF)可抑制pSTAT3+ myMAFs的活化并阻止轉移灶生長
研究人員發現LIF作為癌細胞來源的關鍵因子,與巨噬細胞分泌的顆粒蛋白前體協同作用,通過JAK/STAT通路誘導肝星狀細胞向pSTAT3+ myMAF表型轉化。體外實驗表明,LIF與顆粒蛋白前體共刺激可顯著誘導myMAF特征基因表達并促進癌細胞集落形成,而中和LIF或敲除LIFR可抑制該過程;體內研究也證實,腫瘤相關巨噬細胞和癌細胞分別是顆粒蛋白前體和LIF的主要來源,抑制二者可減少肝轉移灶生長及纖維化。

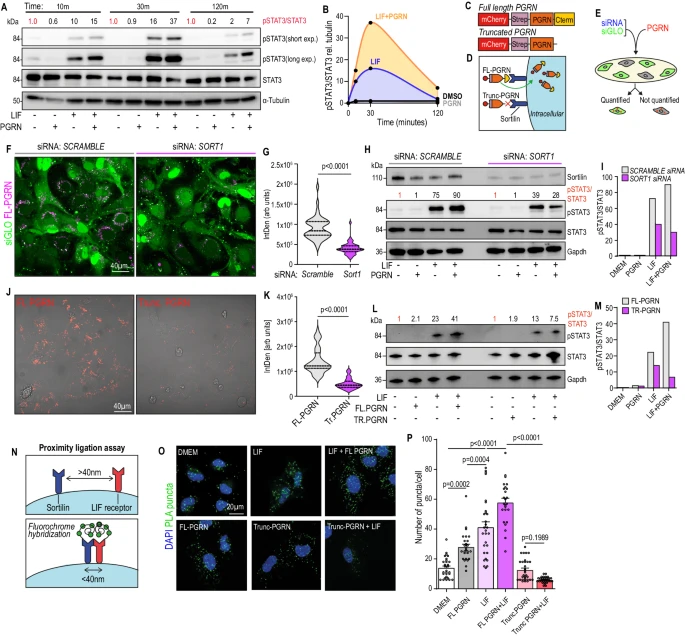
6. 顆粒蛋白前體與Sortilin的結合可增強Sortilin-LIFR-的鄰近性,從而導致肝星狀細胞中STAT3的過度激活
研究人員通過LX2細胞發現顆粒蛋白前體通過C末端與Sortilin受體結合,增強Sortilin與LIFR的鄰近性,從而放大LIF誘導的JAK/STAT信號,促進pSTAT3+ myMAFs表型的誘導,該過程依賴Sortilin介導的顆粒蛋白前體攝取及脂筏聚類介導的信號集中。
7. STAT3激活myMAFs分泌骨橋蛋白(Spp1),而Spp1又繼而支持免疫抑制性巨噬細胞的功能
研究人員在明確pSTAT3+ myMAF活化機制后,探究其促轉移功能,發現myMAF通過分泌骨膜蛋白(Postn)以STAT3依賴方式促進癌細胞增殖,還能分泌骨橋蛋白(OPN)誘導巨噬細胞向免疫抑制表型極化,且癌細胞也可表達OPN協同塑造免疫抑制微環境,此外KPC細胞衍生的巨噬細胞集落刺激因子(mCSF)等其他因子也參與其中,揭示了癌細胞、成纖維細胞和巨噬細胞間的復雜互作促進轉移微環境形成。
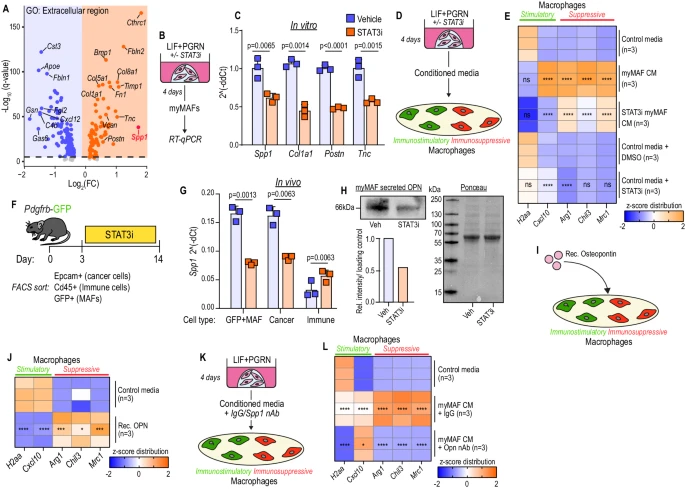
8. myMAF 在轉移性 PDAC 中以 STAT3 依賴性方式編排免疫抑制微環境
研究人員通過STAT3條件性敲除小鼠(STAT3cKO)和STAT3藥理抑制劑(STAT3i)實驗證實,肝星狀細胞中STAT3缺失或抑制可減少免疫抑制性YM1+巨噬細胞,增加CD8+ T細胞浸潤與活化,增強抗腫瘤免疫。機制上,癌細胞與巨噬細胞通過顆粒蛋白前體和LIF激活myMAFs,其分泌的骨膜蛋白促進癌細胞增殖,分泌的骨橋蛋白誘導巨噬細胞向免疫抑制表型極化,形成利于PDAC肝轉移的微環境。

四、 總結
該研究探討了巨噬細胞與纖維細胞之間的JAK/STAT信號通路相互作用如何促進胰腺癌向肝臟的轉移。研究人員發現,這種跨細胞信號傳導在胰腺癌的肝轉移過程中起關鍵作用,尤其是在巨噬細胞和纖維細胞之間的JAK/STAT信號通路激活后,能夠促進腫瘤細胞的轉移和生長。這一發現為理解胰腺癌轉移機制提供了新的視角,并可能為開發針對該過程的治療策略提供潛在靶點。
參考文獻
Raymant M, Astuti Y, Alvaro-Espinosa L, et al. Macrophage-fibroblast JAK/STAT dependent crosstalk promotes liver metastatic outgrowth in pancreatic cancer[J]. Nat Commun, 2024,15(1):3593.
 歡迎來到
歡迎來到 聯系人:張
聯系人:張 地址:上海市長江南路180號長江軟件園B區B637室
地址:上海市長江南路180號長江軟件園B區B637室 郵箱:2844970554@qq.com
郵箱:2844970554@qq.com 傳真:
傳真: